Formal Total Synthesis of Atropurpuran (K. Tanino, 2020)
Atropurpuran (1) represents a complex diterpenoid featuring a pentacyclic skeleton. The research group of Keji Tanino from Hokkaido University was now able to report a racemic, formal total synthesis of this natural product in the Journal of Organic Chemistry. From a retrosynthetic view, they aimed to access atropurpuran from 2 by - in part literature known - late-stage functionalization. The key step to synthesize 2 is the Diels-Alder/RCM cascade delivering the authentic skeleton in just one step from 4 using 3 as an intermediate. The precursor 4 itself should be available from 6-methoxy-1-tetralone (5) through aldol addition.

Figure 1: Retrosynthetic analysis of the formal total synthesis of atropurpuran by the Tanino group.
Synthesis of Key Intermediate 4
As already mentioned the total synthesis started with tetralone 5, which was brominated, and after hydroxylation using Hartwig conditions, protection of the free alcohol gave intermediate 6 as shown in Figure 1. Methenylation in alpha position was achieved in two steps by Mannich reaction with in situ generated imine which delivered a mixture of 7a and b. Amine 7b was easily transformed into 7a by methylation of the amine which led to the subsequent elimination. Then the addition of KCN yielded the desired nitrile 8 in 88% from 6. In the following Mukaiyama aldol addition the Tanino group planned to introduce a C3 unit by coupling with acrolein, however, the dr remained low, the yield remained low with long reaction times (3d, not shown), and later, acrolein was not any more commercially available. Fortunately, the usage of crotonaldehyde led to 9 with good diastereoselectivity in short reaction time (1 h) in good yield (79%). To remove the additional carbon a cross-metathesis with ethylene successfully delivered the terminal double bond and after TES protection 10 was achieved in high yield.
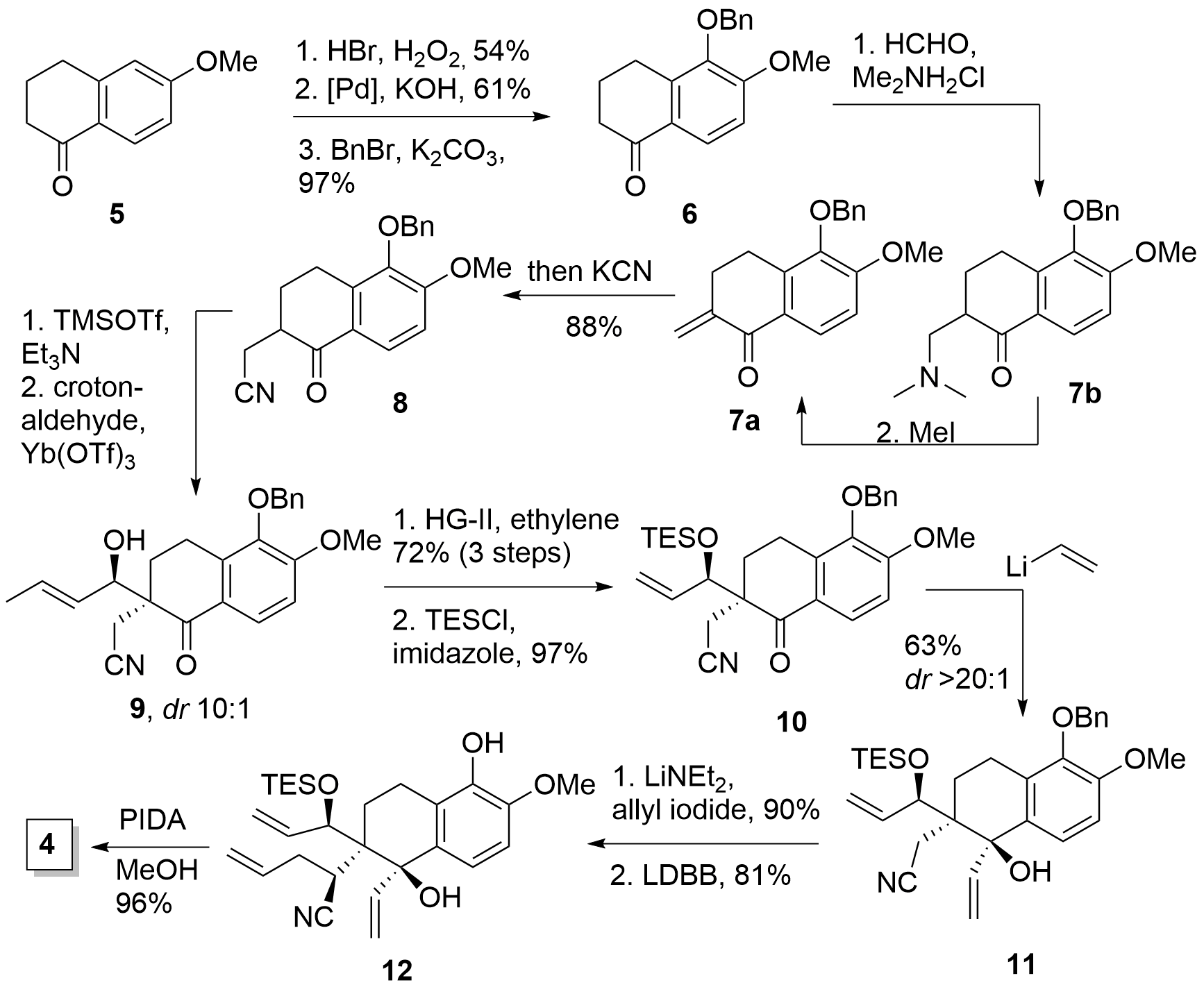
Scheme 1: Synthesis of key intermediate 4.
The addition of vinyl lithium to the carbonyl gave 11 in moderate yield, however in excellent diastereoselectivity. The following addition of allyl iodide was achieved by deprotonation with LiNEt2 which resulted in a single diastereomer. The authors attributed the high selectivity to a chelation effect with the deprotonated alcohol. Finally, removal of the protection group to 12 and PIDA oxidation yielded in 4 as the key intermediate.
Completion of the Formal Total Synthesis
With intermediate 4 in hand, the key Diels Alder reaction was thermally induced and yielded in 3 as a single isomer (Scheme 2). At lower temperatures, HG-II catalyst was added, which led to the ring-closing metathesis to 2. The addition of Pd/C in an H2 atmosphere then led to the reduction of both double bonds. Finally, the addition of thionyl chloride yielded 13 in 56% yield as an impressive one-pot reaction sequence from 4. The authors showed now two different access to key intermediates of previous total syntheses from other groups. One of them is aldehyde 15 which was used before in the total synthesis by Jing Xu published in JACS 2019. To achieve 15 the Tanino group reduced the ketone and the nitrile simultaneously by DIBALH. This was followed by TES deprotection and oxidation to the tricarbonyl 14, which is also literature-known from Xu's total synthesis, however, miss-assigned as the epimer in the alpha position to the aldehyde. The desired intermediate 15 was finally achieved by the alpha methylation using Xu's conditions to complete the formal total synthesis of atropurpuran (1).
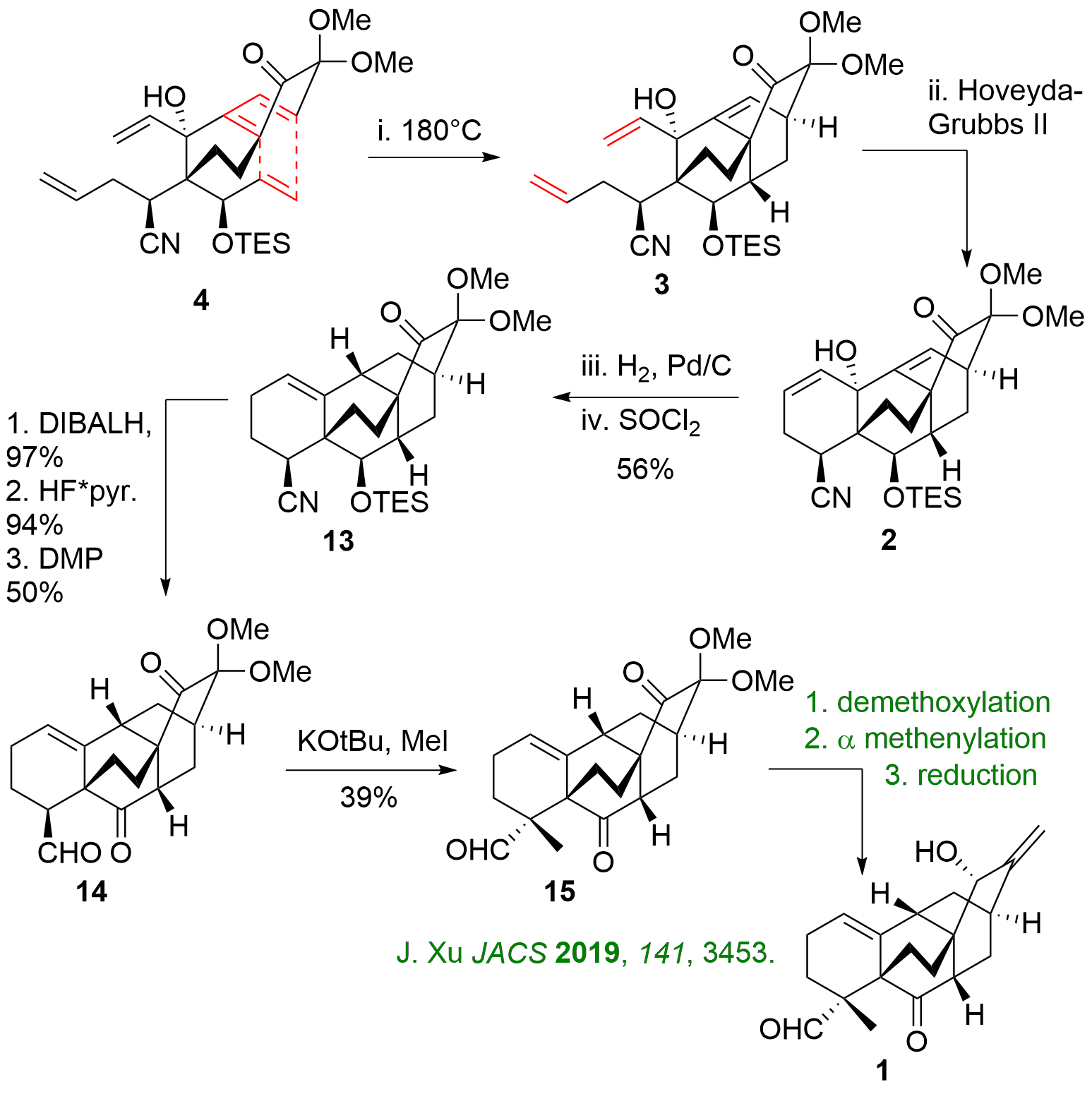
Scheme 2: Completion of the formal total synthesis of atropurpuran (1).
In total, the Tanino group was able to demonstrate a new access to the literature-known intermediate 15 for the total synthesis of atropurpuran (1). Key-step of this synthesis is the one-pot intramolecular Diels Alder reaction/ring-closing metathesis. Furthermore, the Tanino group successfully reported the synthesis of a highly congested intermediate 4, using highly diastereoselective transformations.
Published in T. Suzuki, T. Koyama, K. Nakanishi, S. Kobayashi, K. Tanino Journal of Organic Chemistry 2020, 85, 10125-10135. doi: 10.1021/acs.joc.0c01462
No Comments