Synthesis of Antiviral Nucleoside Analogue Remdesivir
Remdisivir (1) is a nucleoside analogue prodrug of GS-441524 (2, Fig. 1) developed by Gilead Science. In literature, the active metabolite was first described in 2012 by Aesop Cho and coworkersco-workers from Gilead in the journal Bioorganic and Medicinal Chemical Letters and Tetrahedron Letters.[1,2] At that time the antiviral activity against several viruses was demonstrated including HCV, Dengue, Influenza and SARS-CoV.
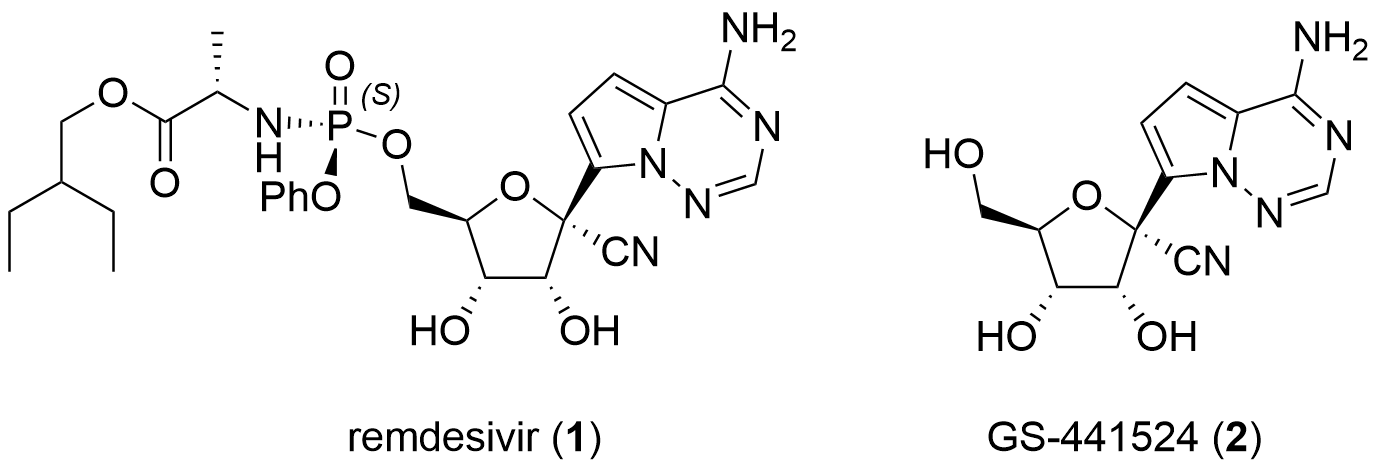
Figure 1: Structure of remdesivir (1) and GS-441524 (2).
Remdesivir (1) and parent compound 2 are adenosine nucleoside analogues. They were modified towards the more stable C-nucleosides and by introducing a cyano group in C1' position. They are also structurally similar to tubercidin, the 7-deaza-adenosine analogue alsowhich can be found in nature. In contrast to 2, remdesivir 1 is coupled with phosphoramidate which leads to improved activity and pharmacokinetic properties. In the following paragraphs, the synthesis of those nucleoside analogues is described. Note, that only publications are included. Synthetic routes from patents were not reviewed.
First reported Synthesis by Gilead (2016, Nature)
Following their initial synthesis of GS-441524 in 2012, Thomas Civlar, Sina Bavari and co-workers from Gilead reported the synthesis of 1 in the journal Nature.[3] The sequence starts with benzylated and oxidized ribose 3 which is coupled with nucleobase 4 in 40% yield. First, the amine of the nucleobase is TMS protected, and then lithium iodide exchange leads to the active lithium organyl species attacking ester 3.3. Product 5 was isolated as an isomeric mixture. Then the free alcohol was transferred into the triflate which was then attacked by the cyanide and after Lewis acidic deprotection GS-441524 (2) was isolated as a pure isomer.

Scheme 1: First-generation synthesis of remdesivir (1).
After the successful synthesis of the active species, the ProTide derivative was targeted. Therefore, the 2',3'-diol was protected using dimethoxypropane (6) under slightly acidic conditions yielding 7 in 90% yield. Coupling of 7 with chiral phosphoramidate 8 using Lewis acidic activation led to 9 in 70% yield. Note, that 8 is attacked under inversion of configuration. Finally, acidic deprotection of 9 yielded in remdesivir (1) in 69%.
Synthesis of Intermediates 3 and 8
Although 3 is commercially available, it can also be prepared in 4 steps from D-ribose (10, see Scheme 2). Jeroen Codée described in 2014[4] the synthesis of protected ribose 11 in 3 steps. First, the formation of methoxide in C1' position, followed by benzylation of the remaining alcohols. Then deprotection to 11 was achieved in 57% using acidic conditions. Finally, Albright-Goldman oxidation gave access to ester 3.[5]
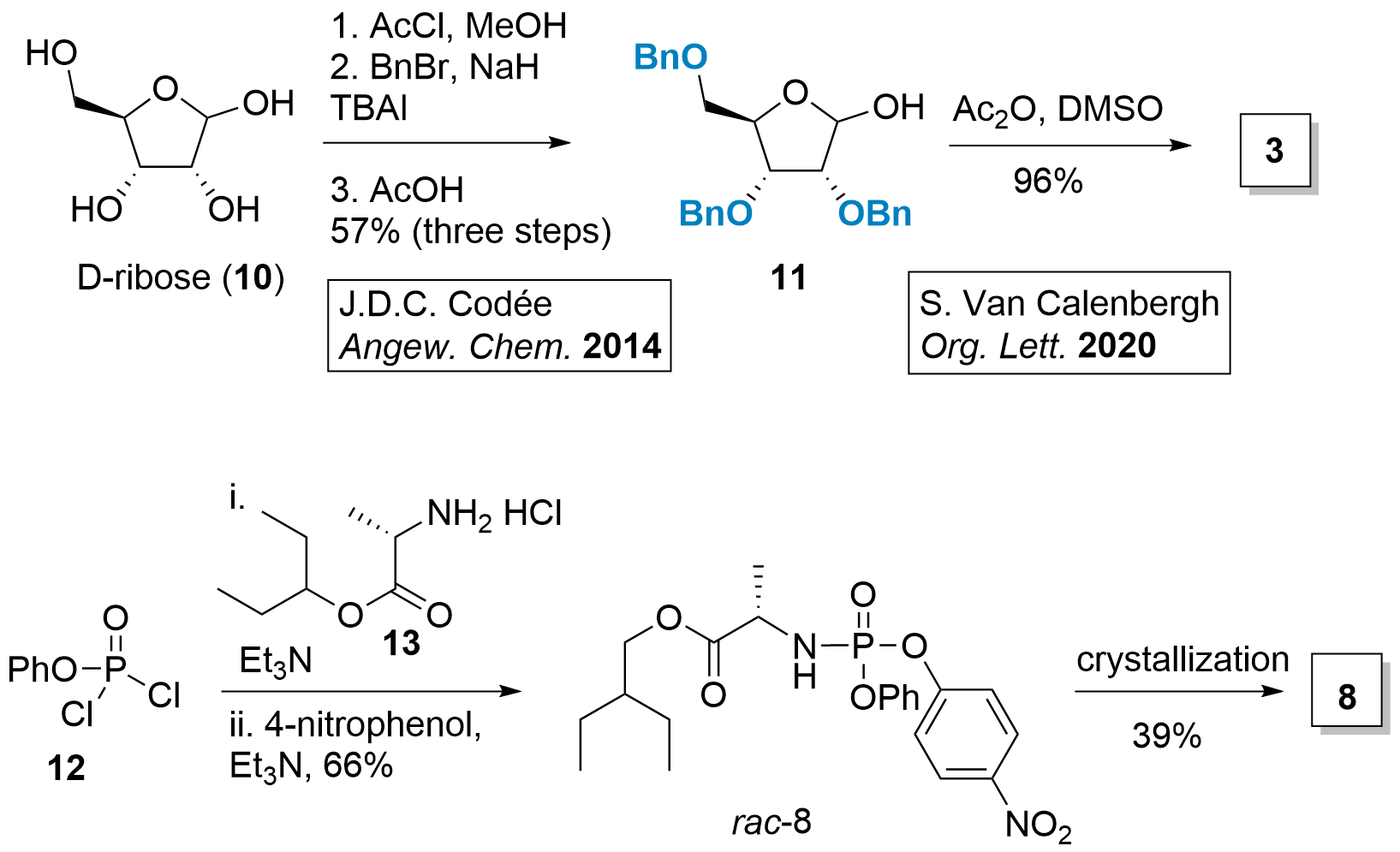
Scheme 2: Synthesis of intermediates 3 and 8.
Phosporamidate 8 can be synthesized from 12 by simple coupling reactions. Racemic 8 is then crystallized to pure 8 for further usage.
Alternative Couplings of Ribose with Nucleobase
To improve the synthetic route, several groups reported improved conditions for coupling the nucleoside base with the ribose sugar core. Two of them will be presented here.
Large-Scale C-Glycosylation by Y. Qin (2020, OPR&D)
In 2020, Yong Qin and co-workers from Sichuan University presented in the journal Organic Process Research & Development an improved coupling strategy with slightly modified conditions (see Scheme 3).[6] First, the free amine wasof 14 as protected using a disilane and Turbothe turbo Grignard reagent (iPrMgCl LiCl) was replaced by nBuLi. Secondary amine iPr2NH should stabilize lithiated 15 and facilitate the protection of 14. With this method, 5 could be synthesized in 75% yield. Importantly, large-scale synthesis of 5 (using 180 g of 14) also gave acceptable results (62% after recrystallization).
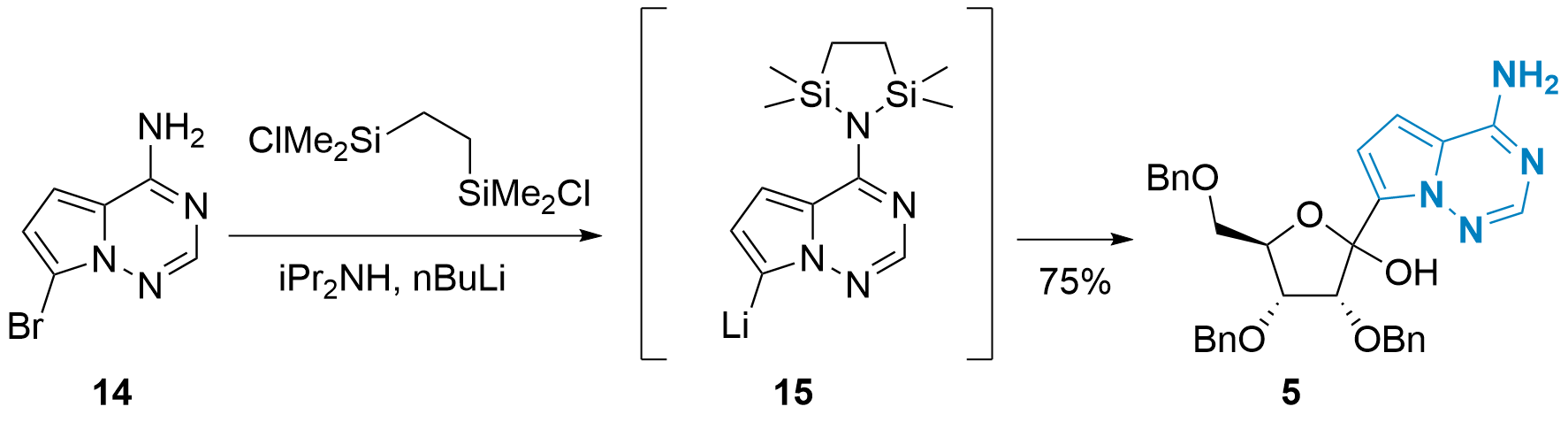
Scheme 3: Improved C-glycosylation by Y. Qin et al.
Direct Glycosylation of Ribose by B. List (2022, Angewandte Chemie)
A completely different approach was reported in Angewandte Chemie by Benjamin List and co-workers from the Max Planck Institut für Kohlenforschung (Mülheim a.d. Ruhr, Germany).[7] As shown in Scheme 4, unprotected ribose 10 was chosen as the starting material. Using catalytic amounts of TMSOTf and BSTFA (N,O-Bis(trimethylsilyl)trifluoroacetamide) as stoichiometric TMS source yielded in opened 17 after arylation with 16 at C1'. In situ TMS deprotection and acetylation then liberated 18 in 60% yield. Note, that the isomeric ratio can be controlled bydepending on the temperature during the HCl treatment.
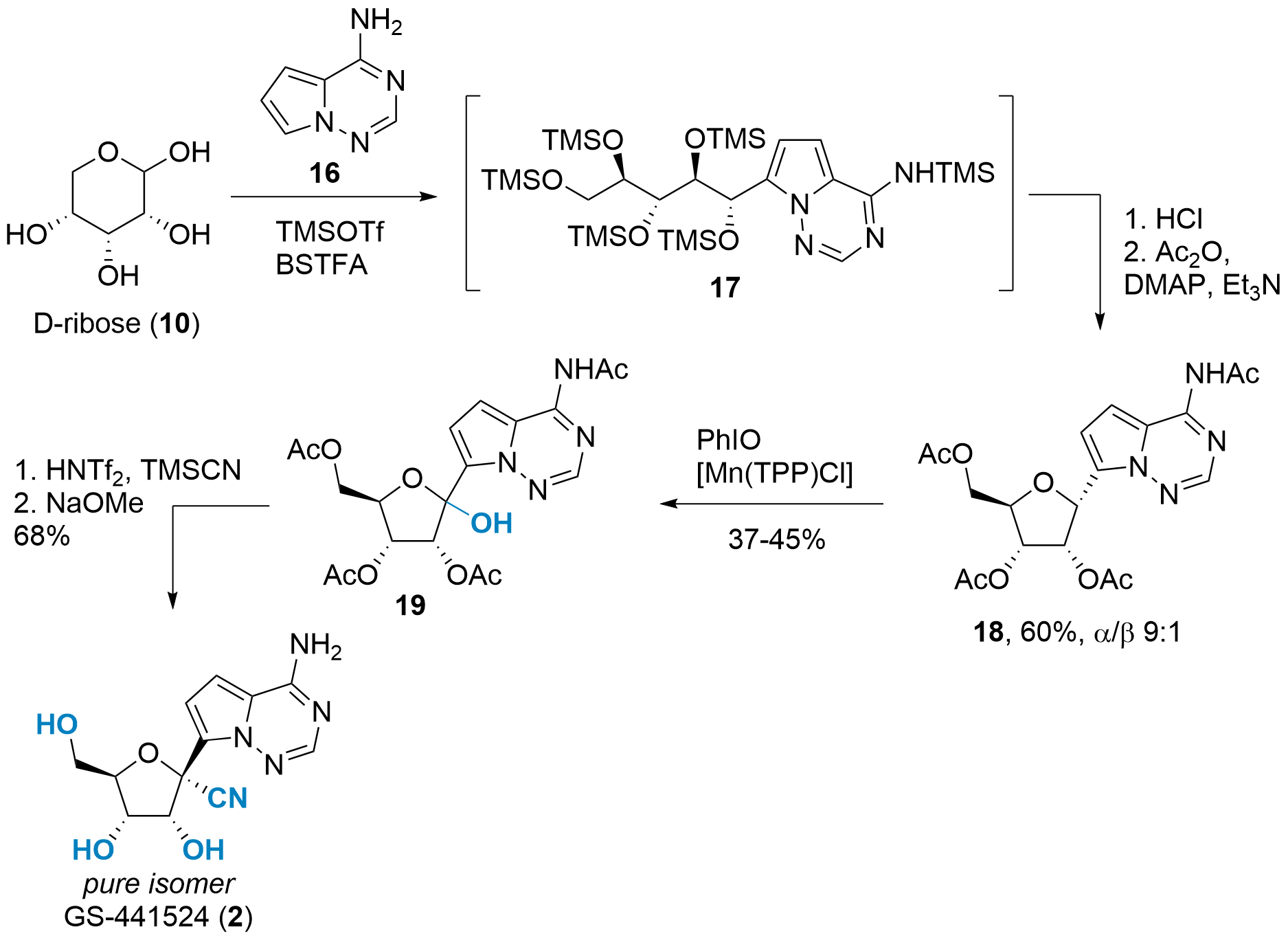
Scheme 4: C-glycosylation of unprotected ribose and oxidation towards GS-441524 (2).
The oxidation of C1' proved to be difficult as overoxidation lead to a low yield. However, intermediate 19 could be isolated using Manganmanganese catalysis and iodosobenzene. The final transformation to GS-441524 was inspired by previous syntheses and yielded 2 in two steps and a 68% yield.
Selective Formation of Phosphoramidate
The usage of chiral 8 limits the usefulness of the synthetic route, therefore, stereoselective coupling reactions with racemic substrates have been developed in the meantime. Two of them are described in Scheme 5. Wanbin Zhang and co-workers from Shanghai Jiao Tong University (China) reported the coupling of P-racemic substrate 20 with intermediate 7 in the journal Angewandte Chemie in 2020.[8] Bicyclic imidazole catalyst 21a first reacts with 20 to active intermediate 22. This intermediate is in equilibrium with its RP-isomer due to substitution by 21a from the other side (not shown), however, it is unfavored by 3.5 kcal/mol. Finally, reaction with 7 gave 9 in 89% yield in excellent dr (>99:1).
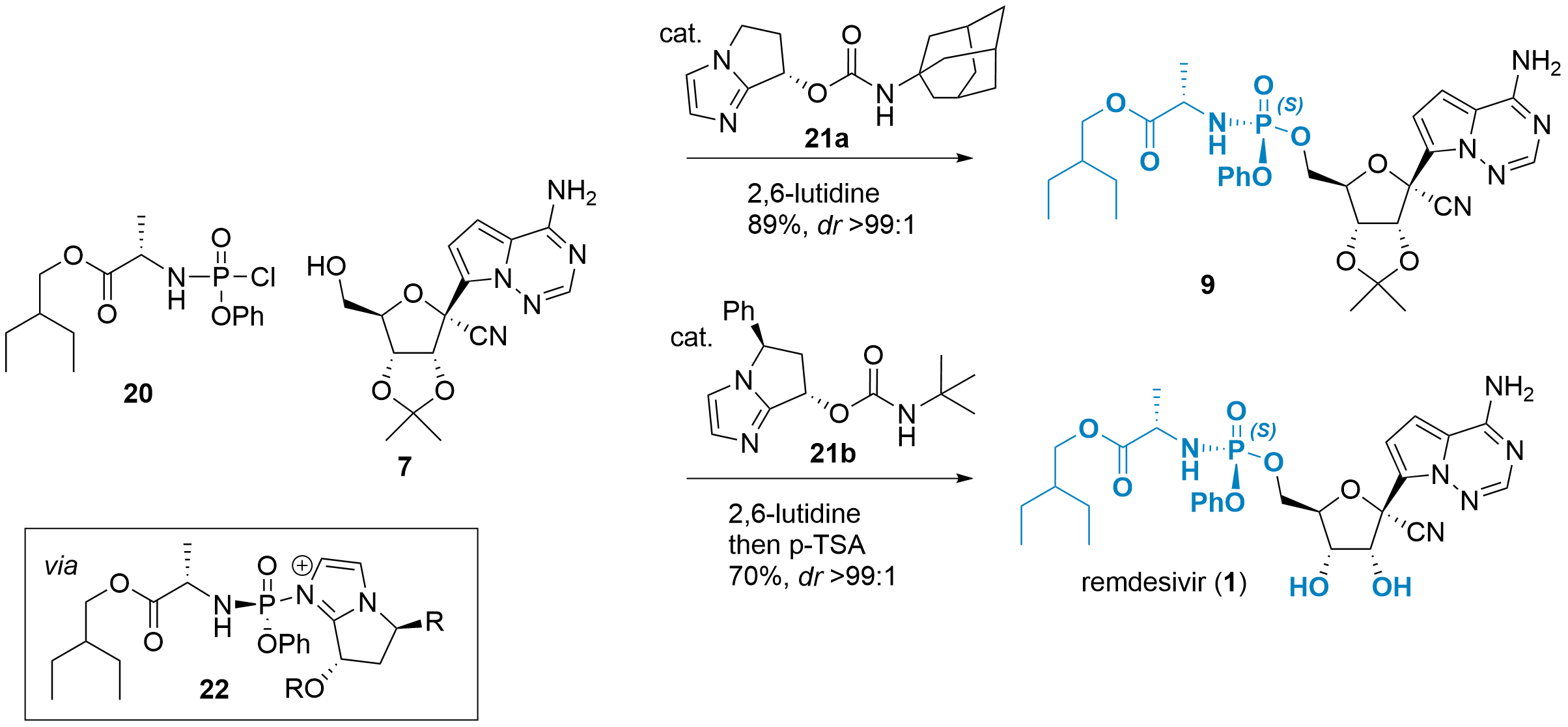
Scheme 5: Usage of catalyst 21 allows the stereoselective phosphoramidate formation.
In a similar report in the Journal of Organic Chemistry from Shang-Cheng Hung and colleagues from the Genomics Research Center (Taipei, Taiwan) catalyst 21b was used with similar efficiency. Note, that the authors decided to directly use a one-pot procedure for the deprotection yielding remdesivir (1) in 70% total yield.
Literature
[1] A. Cho, O. L. Saunders, T. Butler, L. Zhang, J. Xu, J. E. Vela, J. Y. Feng, A. S. Ray, C. U. Kim Synthesis and antiviral activity of a series of 1′-substituted 4-aza-7,9-dideazaadenosine C-nucleosides. Bioorg Med Chem Lett 2012, 22, 2705-2707. https://doi.org/10.1016/j.bmcl.2012.02.105
[2] S. E. Metobo, J. Xu, O. L. Saunders, T. Butler, E. Aktoudianakis, A. Cho, C. U. Kim Practical synthesis of 1′-substituted Tubercidin C-nucleoside analogs. Tetrahedron Lett 2012, 53, 484-486. http://doi.org/10.1016/j.tetlet.2011.11.055
[3] T. Warren, R. Jordan, M. Lo et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. https://doi.org/10.1038/nature17180
[4] E.R. van Rijssel, P. van Delft, G. Lodder, H.S. Overkleeft, G.A. van der Marel, D.V. Filippov, J. D. C. Codée Furanosyl Oxocarbenium Ion Stability and Stereoselectivity Angew Chem Int Ed 2014, 53, 39, 10381-10385. https://doi.org/10.1002/anie.201405477
[5] J. Bouton, S. Van Calenbergh, J. Hullaert Sydnone Ribosides as a Platform for the Synthesis of Pyrazole C-Nucleosides: A Unified Synthesis of Formycin B and Pyrazofurin Org Lett 2020, 22, 23, 9287-9291. https://doi.org/10.1021/acs.orglett.0c03523
[6] F. Xue, Z. Zhou, R. Zhou, X, Zhou, D. Xiao, E. Gu, X. Guo, J. Xiang, K. Wang, L. Yang, W. Zhong, Y. Qin Improvement of the C-glycosylation Step for the Synthesis of Remdesivir Org Process Res Dev 2020, 24, 1772-1777. https://doi.org/10.1021/acs.oprd.0c00310
[7] C. Obradors, B. Mitschke, M. H. Aukland, M. Leutzsch, O. Grossmann, S. Brunen, S. A. Schwengers, B. List Direct and Catalytic C-Glycosylation of Arenes: Expeditious Synthesis of the Remdesivir Nucleoside Angew Chem Int Ed 2022, 61, e202114619. https://doi.org/10.1002/anie.202114619
[8] M. Wang, L. Zhang, X. Huo, Z. Zhang, Q. Yuan, P. Li, J. Chen, Y. Zou, Z. Wu, W. Zhang Catalytic Asymmetric Synthesis of the anti-COVID-19 Drug Remdesivir Angew Chem Int Ed 2020, 59, 20814-20819. http://doi.org/10.1002/anie.202011527
[9] V. Gannedi, B. K. Villuri, S. N. Reddy, C.-C. Ku, C.-H. Wong, S.-C. Hung Practical Remdesivir Synthesis through One-Pot Organocatalyzrf Asymmetric (S)-P-Phosphoramidation J Org Chem 2021, 86, 4997-4985. https://doi.org/10.1021/acs.joc.0c02888